*** LISTEN TO THE PODCAST HERE ***




Zollinger-Ellison Syndrome
Other Known Aliases – Strøm-Zollinger-Ellison Syndrome
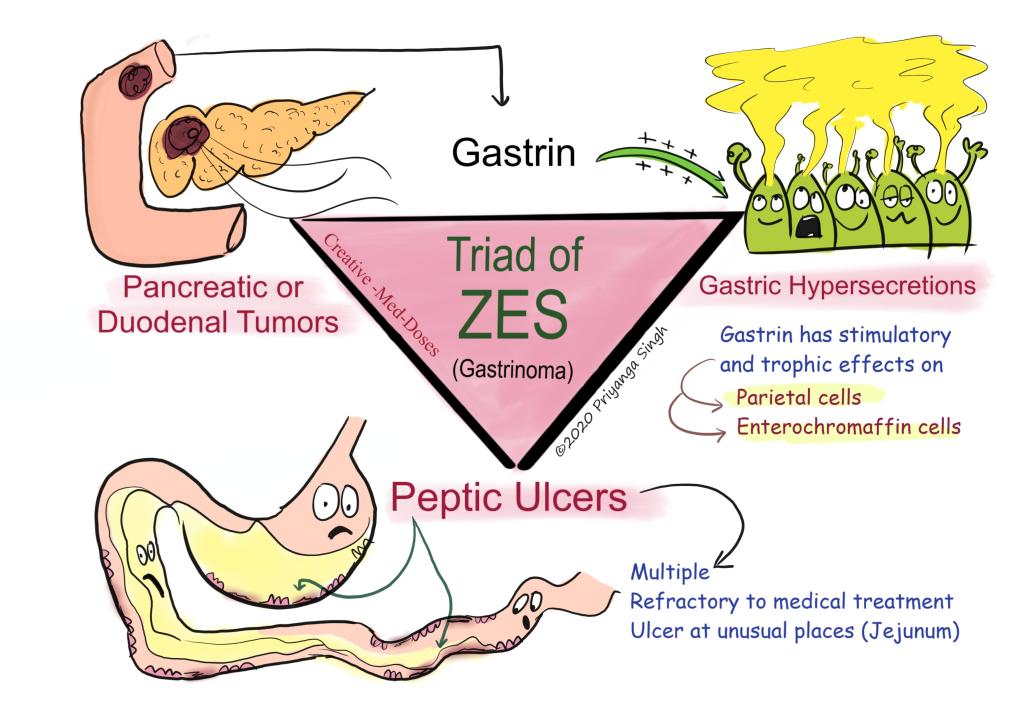
Definition – hypersecretory condition of the stomach caused by a gastrinoma
Clinical Significance – Signs of symptoms of ZES include chronic diarrhea, steatorrhea, GERD, nausea, hematemesis, malabsorption, weight loss, and abdominal pain. This syndrome should be considered in any patient with refractory GERD or PUD. Diagnosis is confirmed with elevated fasting gastrin levels.

History – Named after Robert Milton Zollinger (1903-1992) and Edwin Homer Ellison (1918-1970), who were both American general surgeons and both received their medical doctorates from Ohio State University in 1927 and 1940 respectively. Zollinger would go on to serve in mobile surgical teams during World War II in France and received the Legion of Merit Award and Battle Stars for his contributions. After the war, he would become chairman of the department of surgery at Ohio State in 1947, whee he would spend the majority of career. Ellison would distinguish himself in the realm of research and his career would culminate as chairman of surgery at Marquette School of Medicine in 1967. In 1955, they would present their postulation that hypersecretion of the stomach acid could be due to tumors at the American Surgical Association meeting in Philadelphia and would publish their findings later that year. It should be noted that Roar Strøm, a Norwegian physician, published his findings of this condition in 1952.


References
Castell’s Point
Other Known Aliases – none
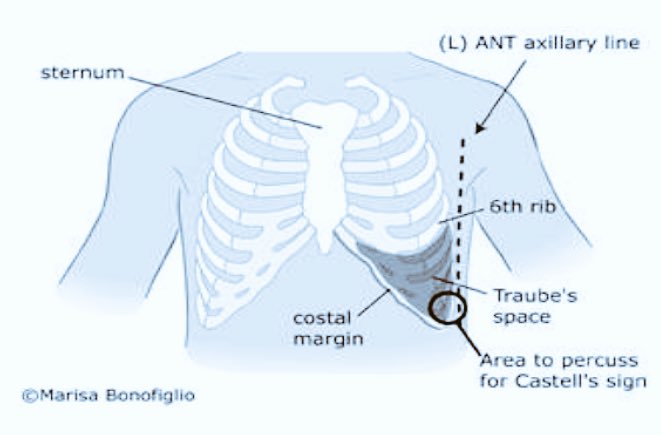
Definition – percussion point on the lowest intercostal space (usually 8th or 9th) in the left anterior axillary line

Clinical Significance – This point is an alternative maneuver to clinically determine if a patient has splenomegaly. With the patient in the supine position, have the patient fully expire and begin percussing in this point while the patient fully inspires. At full expiration, resonance should be appreciated from either the splenic flexure of the colon or gas in the stomach. During inspiration, the spleen moves inferiorly and, if enlarged, will enter this point. This will change the percussion from resonance to dullness.
History – Named after Donald Castell (1935-2021), an American gastroenterologist who received his medical doctorate from George Washington University in 1960. He would spend the majority of his career in the Navy culminating in chair of the Department of Medicine at the National Naval Medical Center in Bethesda, MD. He also held numerous leadership positions national in the American Gastroenterology Association and had a prolific career in scholarship and research. It was through this passion for research that he published his eponymous finding in 1967 while stationed in the Great Lakes naval base in Illinois.


References

***LISTEN TO THE PODCAST HERE***

***LISTEN TO THE PODCAST HERE***
Traube’s Space
Other Known Aliases – none
Definition – crescent-shaped, anatomic space of the LUQ bordered by the lower edge of the lung, anterior border of the spleen, left costal margin, and the inferior margin of the left lobe of the liver
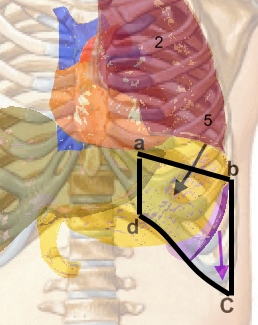
Clinical Significance – Clinically, the surface borders are the sixth rib superiorly, the left mid-axillary line laterally, and the left costal margin inferiorly. The importance of this space is during percussion for splenomegaly. If the spleen is not enlarged, then there will be resonance to percussion. If splenomegaly is present, then there will be dullness. False positives include recent meals, fundal mass, left pleural effusions, or pericardial effusion.
History – Named after Ludwig Traube (1818-1876), a German physician who received his medical doctorate from the University of Berlin in 1840. Due to prejudice against his Jewish ancestry, his academic career was handicapped despite working with notable physicians in Germany and publishing on ground breaking experimental physiology studies, which included using temperature measurement as a routine clinical examination method. A master in auscultation and percussion, he was sought after throughout Germany to study under and utilized these techniques in his popular patient clinics. It was here that one of his former students, Oscar Fraentzel, observed the master clinician and published a report in 1868 on this eponymous space, which he named after his mentor.


References

Question
A 37yo man is presenting to your practice to establish primary care. He reports a history of controlled inflammatory bowel disease and sees a gastroenterologist every 6 months. Before you begin your physical examination:
Answer
A simple mnemonic for the extraintestinal manifestations of inflammatory bowel disease is A PIE SACK:

Question
A 37yo man is presenting to your practice to establish primary care. He reports a history of controlled inflammatory bowel disease and sees a gastroenterologist every 6 months. Before you begin your physical examination:

Puestow Procedure
Other Known Aliases – Puestow-Gillesby procedure, lateral pancreaticojejunostomy
Definition – side-to-side anastomosis of the main pancreatic duct of Wirsung to the proximal jejunum

Clinical Significance – this is a surgical management option for patients with chronic pancreatitis by simultaneously facilitating drainage and preserving physiologic function of the pancreas.
History – Named after Charles Bernard Puestow (1902-1973), an American surgeon who recieved his medical doctorate from the University of Pennsylvania in 1925. He would serve as a military surgeon during the 2nd World War and commanded the 27th Evacuation Hospital providing surgical services to wounded soldiers in Europe and North Africa. His commitment to the veteran population would continue after the war when he established the first surgical residency program based in a veterans hospitals in the United States in 1946. It was at Hines Veterans Hospital in Illinois where he and his partner, William Gillesby, would publish their experience and outcomes on 21 patients with chronic pancreatitis in 1958, which would lead to the creation of his eponymonic surgical procedure.

References

